 Protein Structure 1
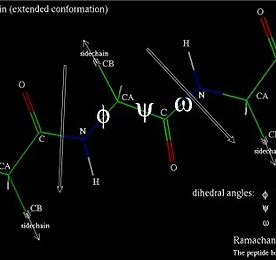
단백질(protein)은 부사슬(sidechain)에 어떤 반복되는 유닛이 결합한 백본(backbone)의 중합체(polymers)라 할 수 있다. 즉, 간단히 말해 다음과 같은 구조를 가진다. 여기서 가장 중심이 되는 분자는 N, Cα, C이며, Cα에 붙은 잔기를 부사슬이라 한다. 단백질의 크기는, H가 하나로만 구성되어 있는 Gly에서부터, 커다란 벤젠 고리(benzeng ring)를 형성하는 Phe까지 매우 다양하다. 또, +, -과 같은 전하를 띄며, 다른 극성을 띄는 잔기와 수소 결합(hydrogen bonds)을 형성하기도 한다. 여기서 hydrophobic이란 물과 친화성이 매우 떨어지는 것을 의미하며, hydrophilic은 물과의 친화성이 높다는 것을 의미한다. 또, 단백질의 모양과 단..
더보기
Protein Structure 1
단백질(protein)은 부사슬(sidechain)에 어떤 반복되는 유닛이 결합한 백본(backbone)의 중합체(polymers)라 할 수 있다. 즉, 간단히 말해 다음과 같은 구조를 가진다. 여기서 가장 중심이 되는 분자는 N, Cα, C이며, Cα에 붙은 잔기를 부사슬이라 한다. 단백질의 크기는, H가 하나로만 구성되어 있는 Gly에서부터, 커다란 벤젠 고리(benzeng ring)를 형성하는 Phe까지 매우 다양하다. 또, +, -과 같은 전하를 띄며, 다른 극성을 띄는 잔기와 수소 결합(hydrogen bonds)을 형성하기도 한다. 여기서 hydrophobic이란 물과 친화성이 매우 떨어지는 것을 의미하며, hydrophilic은 물과의 친화성이 높다는 것을 의미한다. 또, 단백질의 모양과 단..
더보기
 Gibbs Sampling
모티프(Motif)란 DNA, RNA, Protein 어떤 것의 조각이든, 비교적 반복적으로 나타나는 아주 짧은 서열 조각을 말한다. Regulatory Motif의 특징으로, 작고(tiny), 변화가 심하며(highly variable), 서로 거의 비슷한 크기에(constant size) 아주 빈번하게 나타난다. 이 작은 서열 조각은, 다중 서열 정렬을 하는데 아주 중요한 역할을 한다. 일반적으로, 다중 서열 정렬에 관한 문제는 다이나믹 프로그래밍(Dynamic Programming)을 적용하더라도 매우 어려운 문제로 알려져 있다. 어느 정도의 시간이 필요한지, 이것을 효과적으로 계산할 수 있는 다항시간 내의 알고리즘이 존재하는지 여부조차 알 수 없는, NP 문제에 속한다. 그런데, 모티프를 이용하면..
더보기
Gibbs Sampling
모티프(Motif)란 DNA, RNA, Protein 어떤 것의 조각이든, 비교적 반복적으로 나타나는 아주 짧은 서열 조각을 말한다. Regulatory Motif의 특징으로, 작고(tiny), 변화가 심하며(highly variable), 서로 거의 비슷한 크기에(constant size) 아주 빈번하게 나타난다. 이 작은 서열 조각은, 다중 서열 정렬을 하는데 아주 중요한 역할을 한다. 일반적으로, 다중 서열 정렬에 관한 문제는 다이나믹 프로그래밍(Dynamic Programming)을 적용하더라도 매우 어려운 문제로 알려져 있다. 어느 정도의 시간이 필요한지, 이것을 효과적으로 계산할 수 있는 다항시간 내의 알고리즘이 존재하는지 여부조차 알 수 없는, NP 문제에 속한다. 그런데, 모티프를 이용하면..
더보기






